geom_range() and geom_half_range() draw tiles that are designed to
represent range-based genomic features, such as exons. In combination with
geom_intron(), these geoms form the core components for visualizing
transcript structures.
Usage
geom_range(
mapping = NULL,
data = NULL,
stat = "identity",
position = "identity",
...,
vjust = NULL,
linejoin = "mitre",
na.rm = FALSE,
show.legend = NA,
inherit.aes = TRUE
)
geom_half_range(
mapping = NULL,
data = NULL,
stat = "identity",
position = "identity",
...,
range.orientation = "bottom",
linejoin = "mitre",
na.rm = FALSE,
show.legend = NA,
inherit.aes = TRUE
)Arguments
- mapping
Set of aesthetic mappings created by
aes(). If specified andinherit.aes = TRUE(the default), it is combined with the default mapping at the top level of the plot. You must supplymappingif there is no plot mapping.- data
The data to be displayed in this layer. There are three options:
If
NULL, the default, the data is inherited from the plot data as specified in the call toggplot().A
data.frame, or other object, will override the plot data. All objects will be fortified to produce a data frame. Seefortify()for which variables will be created.A
functionwill be called with a single argument, the plot data. The return value must be adata.frame, and will be used as the layer data. Afunctioncan be created from aformula(e.g.~ head(.x, 10)).- stat
The statistical transformation to use on the data for this layer. When using a
geom_*()function to construct a layer, thestatargument can be used the override the default coupling between geoms and stats. Thestatargument accepts the following:A
Statggproto subclass, for exampleStatCount.A string naming the stat. To give the stat as a string, strip the function name of the
stat_prefix. For example, to usestat_count(), give the stat as"count".For more information and other ways to specify the stat, see the layer stat documentation.
- position
A position adjustment to use on the data for this layer. This can be used in various ways, including to prevent overplotting and improving the display. The
positionargument accepts the following:The result of calling a position function, such as
position_jitter(). This method allows for passing extra arguments to the position.A string naming the position adjustment. To give the position as a string, strip the function name of the
position_prefix. For example, to useposition_jitter(), give the position as"jitter".For more information and other ways to specify the position, see the layer position documentation.
- ...
Other arguments passed on to
layer()'sparamsargument. These arguments broadly fall into one of 4 categories below. Notably, further arguments to thepositionargument, or aesthetics that are required can not be passed through.... Unknown arguments that are not part of the 4 categories below are ignored.Static aesthetics that are not mapped to a scale, but are at a fixed value and apply to the layer as a whole. For example,
colour = "red"orlinewidth = 3. The geom's documentation has an Aesthetics section that lists the available options. The 'required' aesthetics cannot be passed on to theparams. Please note that while passing unmapped aesthetics as vectors is technically possible, the order and required length is not guaranteed to be parallel to the input data.When constructing a layer using a
stat_*()function, the...argument can be used to pass on parameters to thegeompart of the layer. An example of this isstat_density(geom = "area", outline.type = "both"). The geom's documentation lists which parameters it can accept.Inversely, when constructing a layer using a
geom_*()function, the...argument can be used to pass on parameters to thestatpart of the layer. An example of this isgeom_area(stat = "density", adjust = 0.5). The stat's documentation lists which parameters it can accept.The
key_glyphargument oflayer()may also be passed on through.... This can be one of the functions described as key glyphs, to change the display of the layer in the legend.
- vjust
A numeric vector specifying vertical justification. If specified, overrides the
justsetting.- linejoin
Line join style (round, mitre, bevel).
- na.rm
If
FALSE, the default, missing values are removed with a warning. IfTRUE, missing values are silently removed.- show.legend
logical. Should this layer be included in the legends?
NA, the default, includes if any aesthetics are mapped.FALSEnever includes, andTRUEalways includes. It can also be a named logical vector to finely select the aesthetics to display.- inherit.aes
If
FALSE, overrides the default aesthetics, rather than combining with them. This is most useful for helper functions that define both data and aesthetics and shouldn't inherit behaviour from the default plot specification, e.g.borders().- range.orientation
character()one of "top" or "bottom", specifying where the half ranges will be plotted with respect to each transcript (y).
Value
the return value of a geom_* function is not intended to be
directly handled by users. Therefore, geom_* functions should never be
executed in isolation, rather used in combination with a
ggplot2::ggplot() call.
Details
geom_range() and geom_half_range() require the following aes();
xstart, xend and y (e.g. transcript name). geom_half_range() takes
advantage of the vertical symmetry of transcript annotation by plotting only
half of a range on the top or bottom of a transcript structure. This can be
useful for comparing between two transcripts or free up plotting space for
other transcript annotations (e.g. geom_junction()).
Examples
library(magrittr)
library(ggplot2)
# to illustrate the package's functionality
# ggtranscript includes example transcript annotation
sod1_annotation %>% head()
#> # A tibble: 6 × 8
#> seqnames start end strand type gene_name transcript_name
#> <fct> <int> <int> <fct> <fct> <chr> <chr>
#> 1 21 31659666 31668931 + gene SOD1 NA
#> 2 21 31659666 31668931 + transcript SOD1 SOD1-202
#> 3 21 31659666 31659784 + exon SOD1 SOD1-202
#> 4 21 31659770 31659784 + CDS SOD1 SOD1-202
#> 5 21 31659770 31659772 + start_codon SOD1 SOD1-202
#> 6 21 31663790 31663886 + exon SOD1 SOD1-202
#> # ℹ 1 more variable: transcript_biotype <chr>
# extract exons
sod1_exons <- sod1_annotation %>% dplyr::filter(type == "exon")
sod1_exons %>% head()
#> # A tibble: 6 × 8
#> seqnames start end strand type gene_name transcript_name
#> <fct> <int> <int> <fct> <fct> <chr> <chr>
#> 1 21 31659666 31659784 + exon SOD1 SOD1-202
#> 2 21 31663790 31663886 + exon SOD1 SOD1-202
#> 3 21 31666449 31666518 + exon SOD1 SOD1-202
#> 4 21 31667258 31667375 + exon SOD1 SOD1-202
#> 5 21 31668471 31668931 + exon SOD1 SOD1-202
#> 6 21 31659693 31659841 + exon SOD1 SOD1-204
#> # ℹ 1 more variable: transcript_biotype <chr>
base <- sod1_exons %>%
ggplot(aes(
xstart = start,
xend = end,
y = transcript_name
))
# geom_range() is designed to visualise range-based annotation such as exons
base + geom_range()
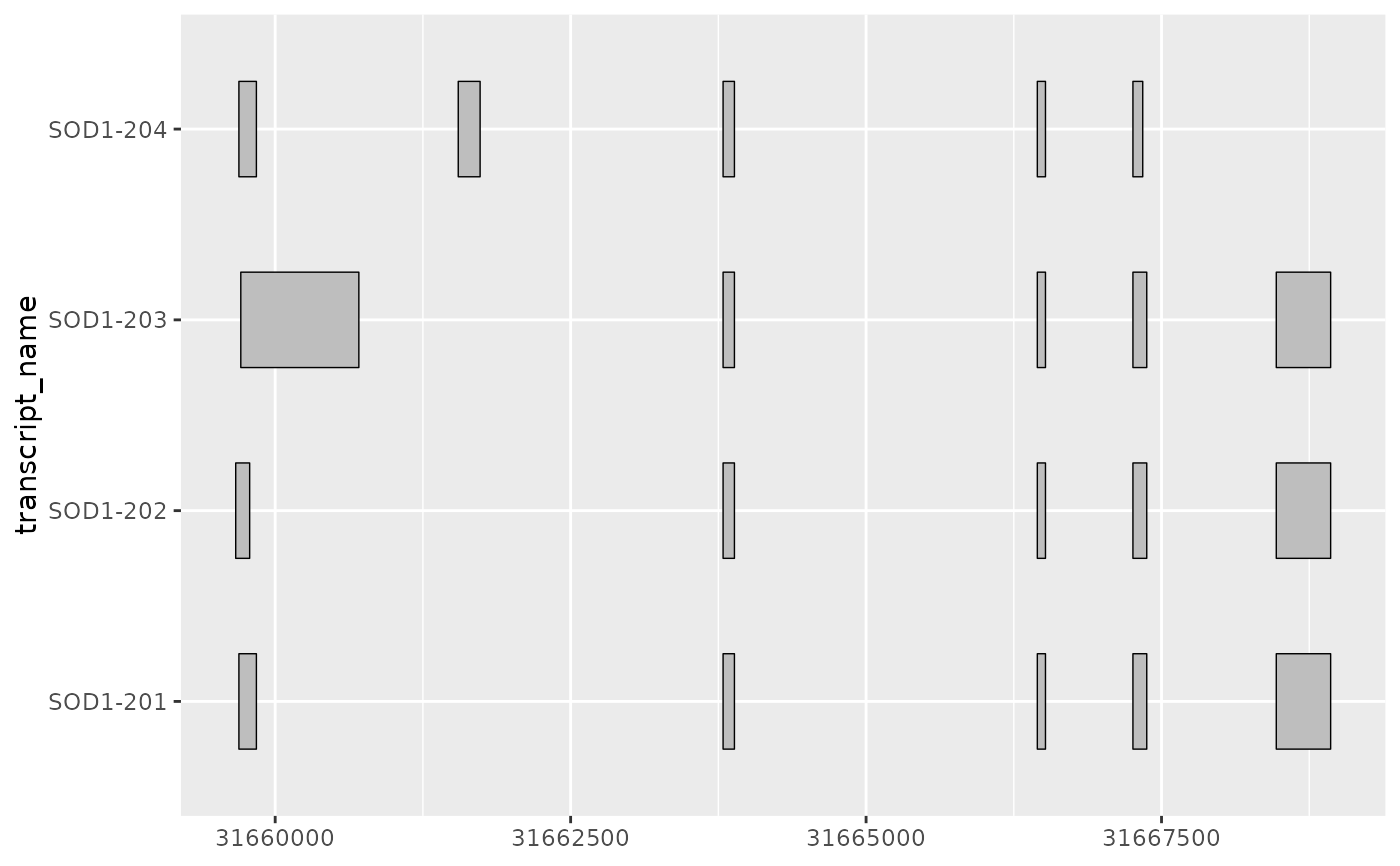 # geom_half_range() allows users to plot half ranges
# on the top or bottom of the transcript
base + geom_half_range()
# geom_half_range() allows users to plot half ranges
# on the top or bottom of the transcript
base + geom_half_range()
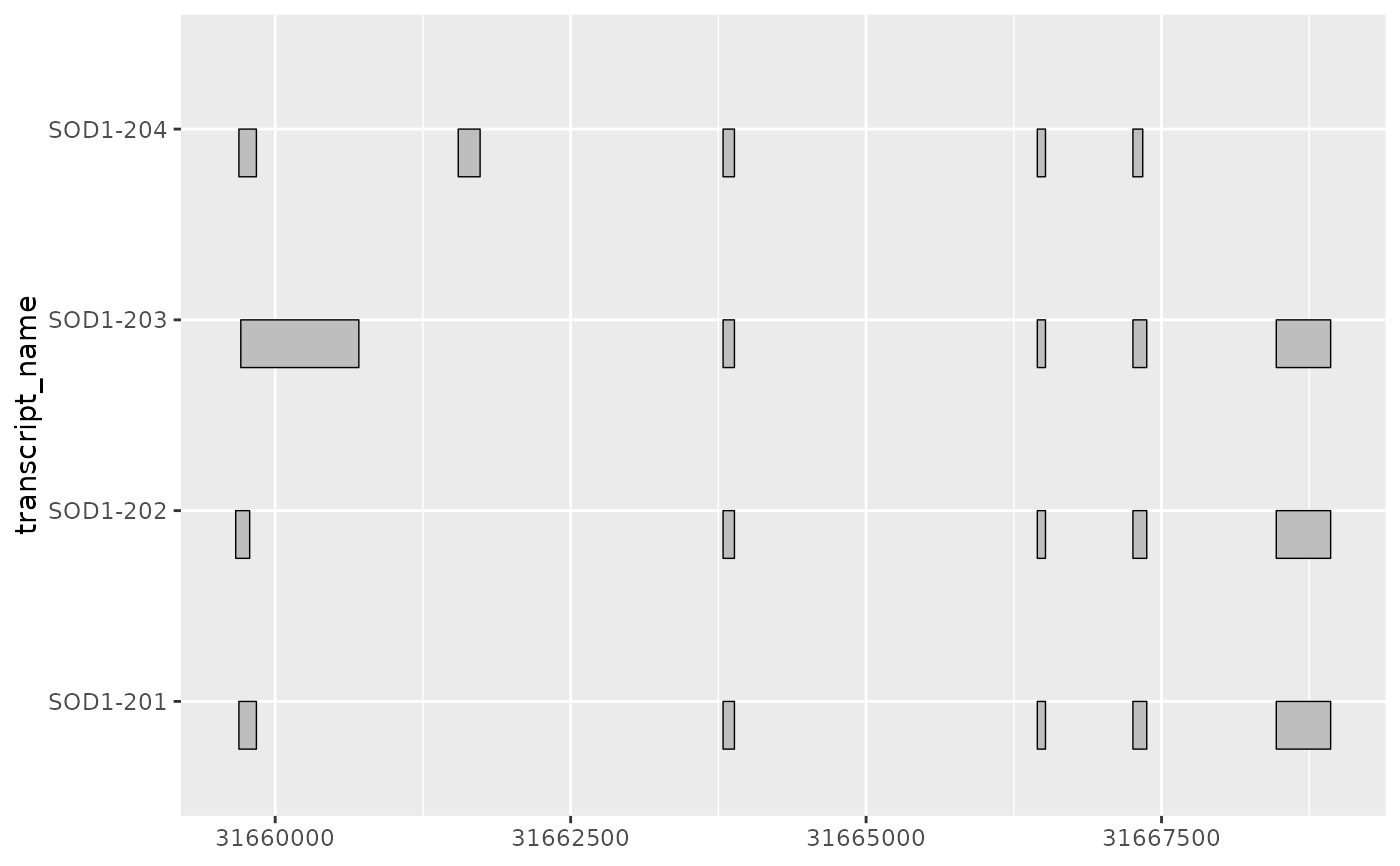 # where the half ranges are plotted can be adjusted using range.orientation
base + geom_half_range(range.orientation = "top")
# where the half ranges are plotted can be adjusted using range.orientation
base + geom_half_range(range.orientation = "top")
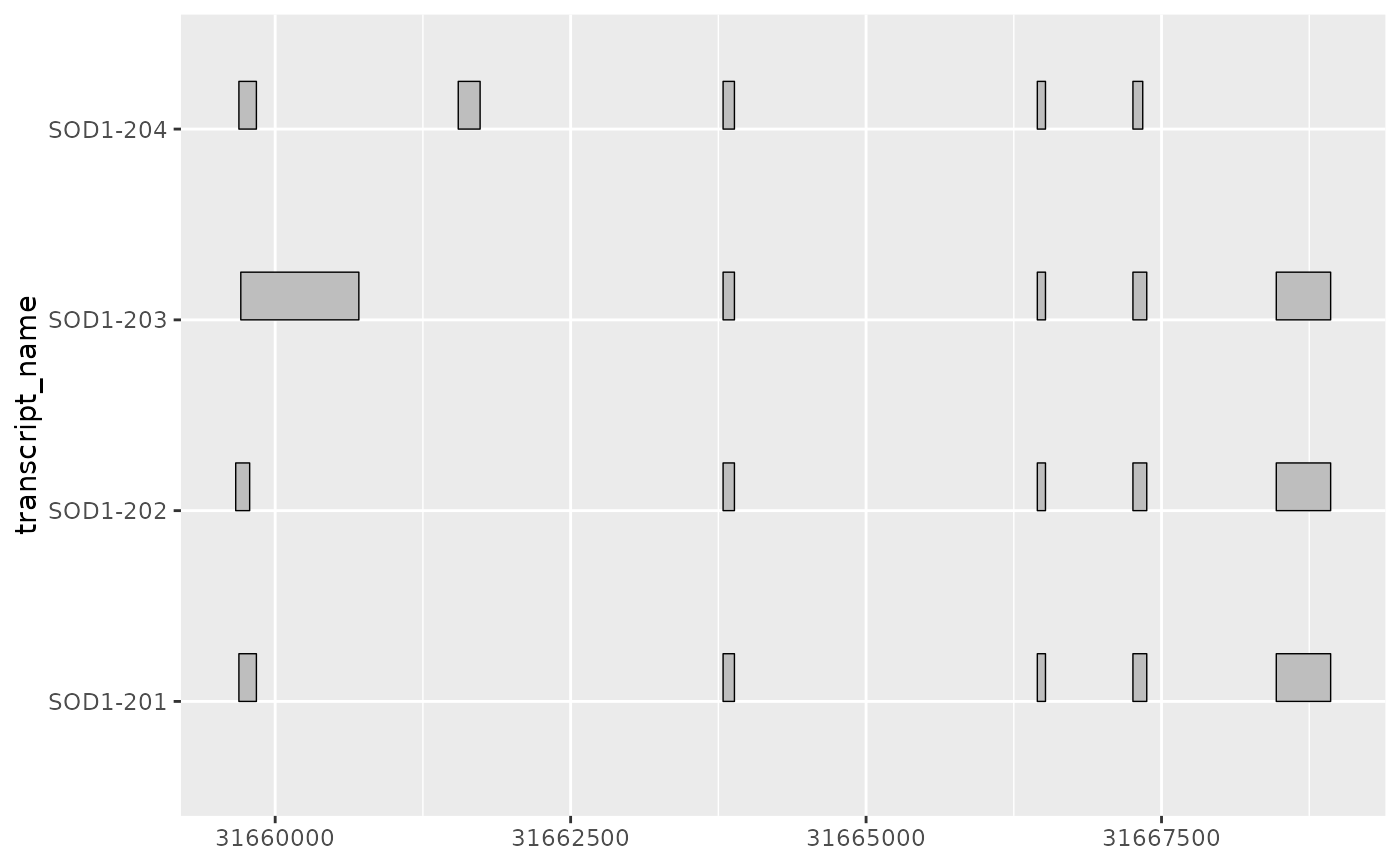 # as a ggplot2 extension, ggtranscript geoms inherit the
# the functionality from the parameters and aesthetics in ggplot2
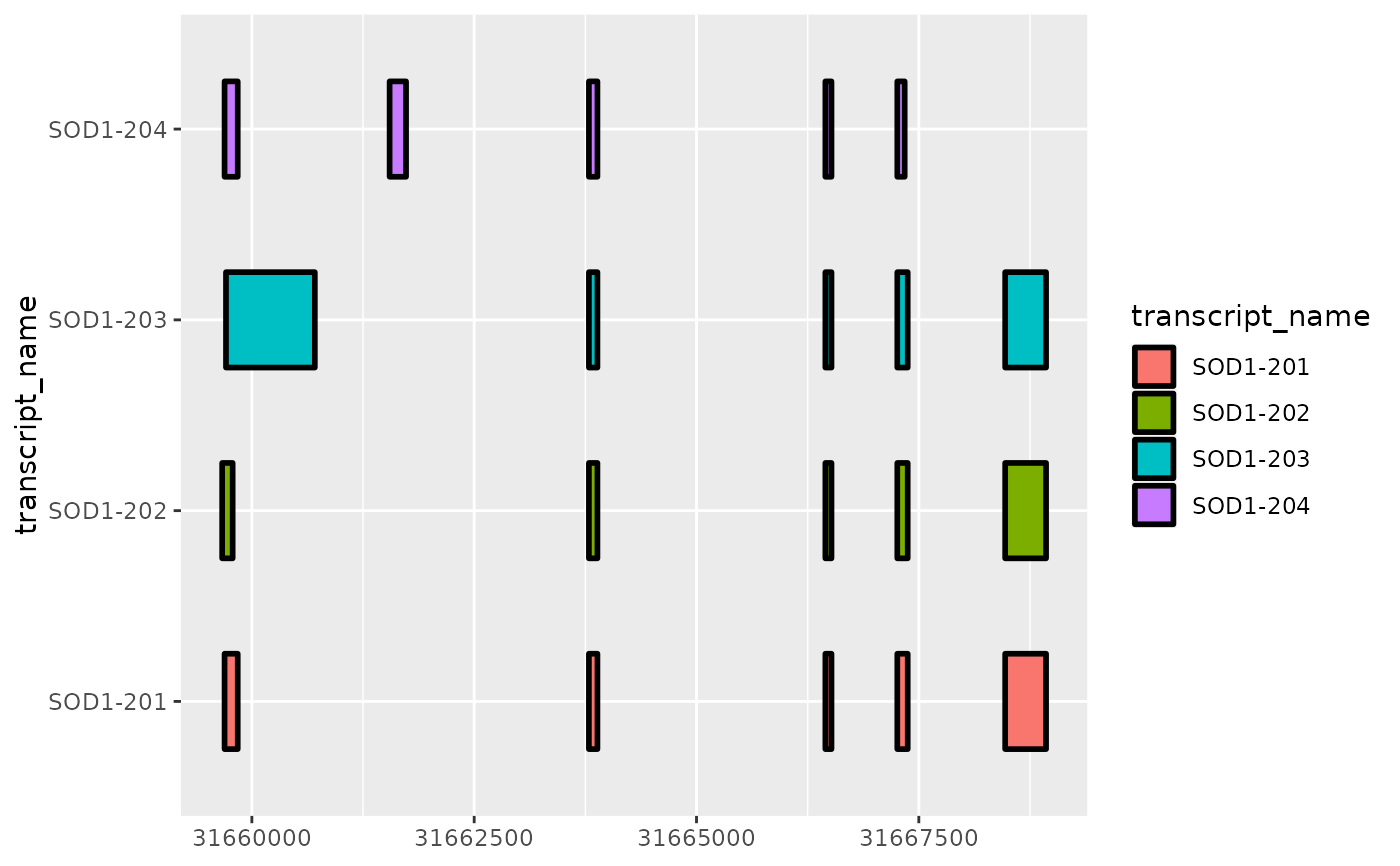
base + geom_range(
aes(fill = transcript_name),
linewidth = 1
)
# as a ggplot2 extension, ggtranscript geoms inherit the
# the functionality from the parameters and aesthetics in ggplot2
base + geom_range(
aes(fill = transcript_name),
linewidth = 1
)
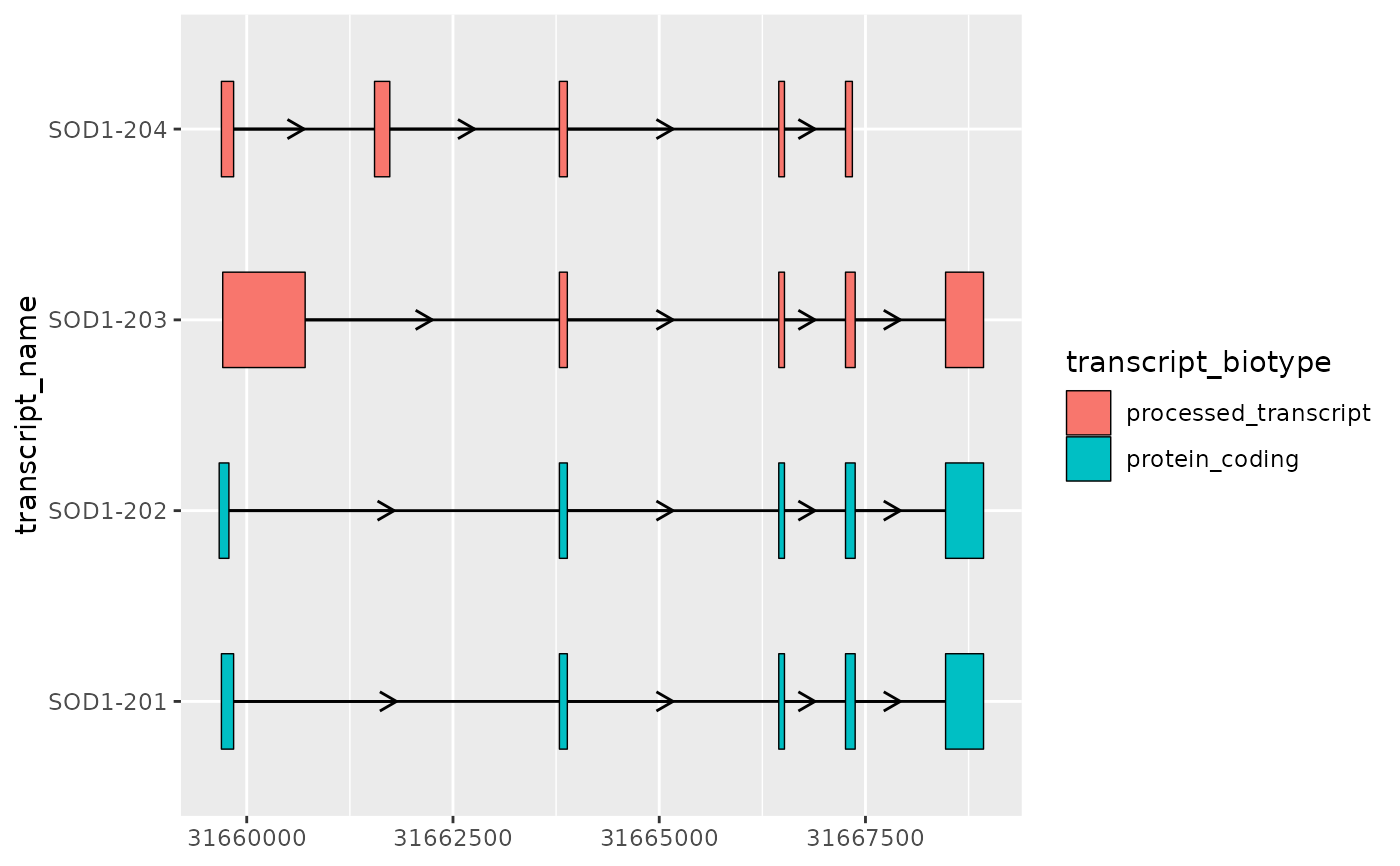
 # together, geom_range() and geom_intron() are designed to visualize
# the core components of transcript annotation
base + geom_range(
aes(fill = transcript_biotype)
) +
geom_intron(
data = to_intron(sod1_exons, "transcript_name")
)
# together, geom_range() and geom_intron() are designed to visualize
# the core components of transcript annotation
base + geom_range(
aes(fill = transcript_biotype)
) +
geom_intron(
data = to_intron(sod1_exons, "transcript_name")
)
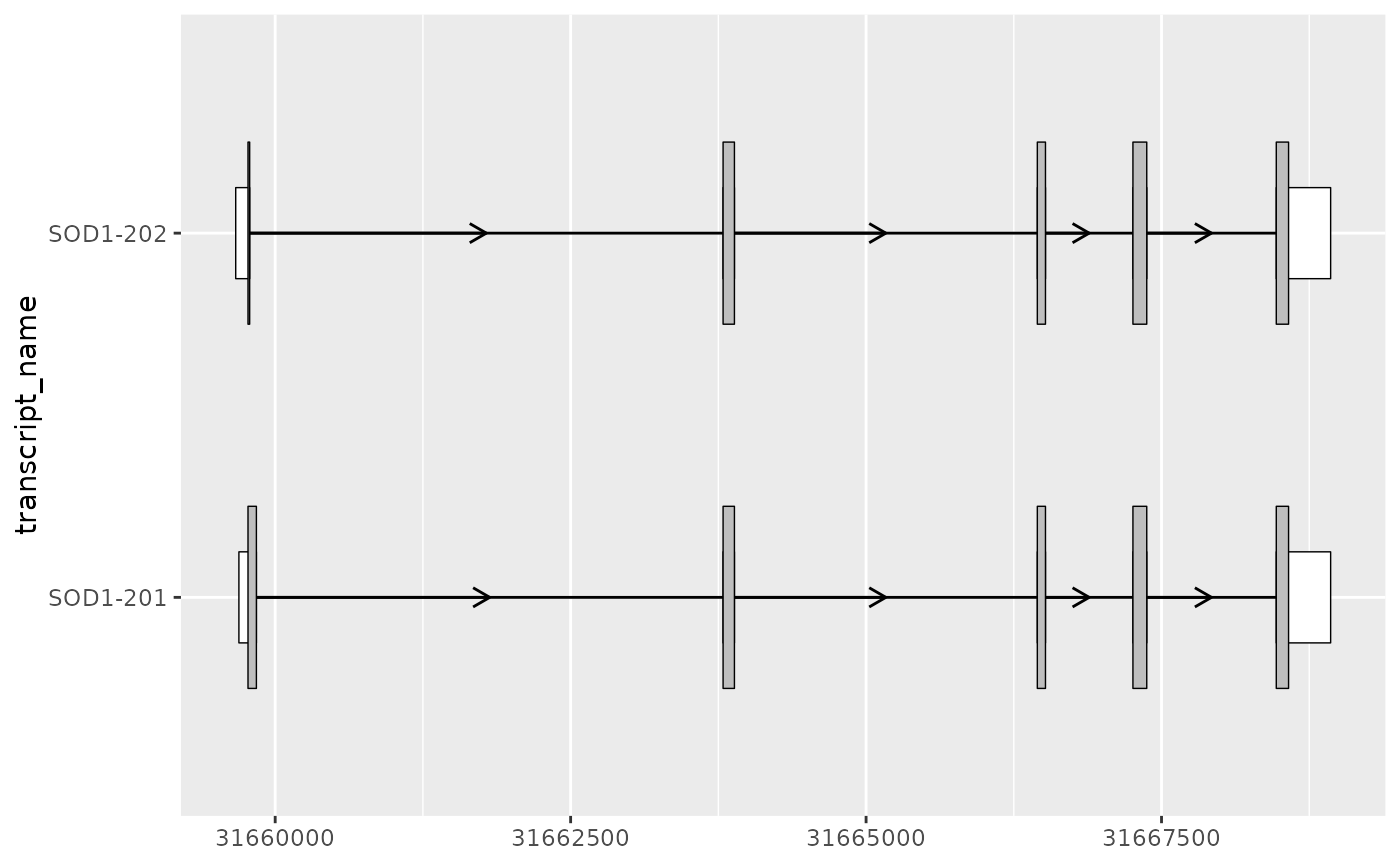
 # for protein coding transcripts
# geom_range() be useful for visualizing UTRs that lie outside of the CDS
sod1_exons_prot_coding <- sod1_exons %>%
dplyr::filter(transcript_biotype == "protein_coding")
# extract cds
sod1_cds <- sod1_annotation %>%
dplyr::filter(type == "CDS")
sod1_exons_prot_coding %>%
ggplot(aes(
xstart = start,
xend = end,
y = transcript_name
)) +
geom_range(
fill = "white",
height = 0.25
) +
geom_range(
data = sod1_cds
) +
geom_intron(
data = to_intron(sod1_exons_prot_coding, "transcript_name")
)
# for protein coding transcripts
# geom_range() be useful for visualizing UTRs that lie outside of the CDS
sod1_exons_prot_coding <- sod1_exons %>%
dplyr::filter(transcript_biotype == "protein_coding")
# extract cds
sod1_cds <- sod1_annotation %>%
dplyr::filter(type == "CDS")
sod1_exons_prot_coding %>%
ggplot(aes(
xstart = start,
xend = end,
y = transcript_name
)) +
geom_range(
fill = "white",
height = 0.25
) +
geom_range(
data = sod1_cds
) +
geom_intron(
data = to_intron(sod1_exons_prot_coding, "transcript_name")
)
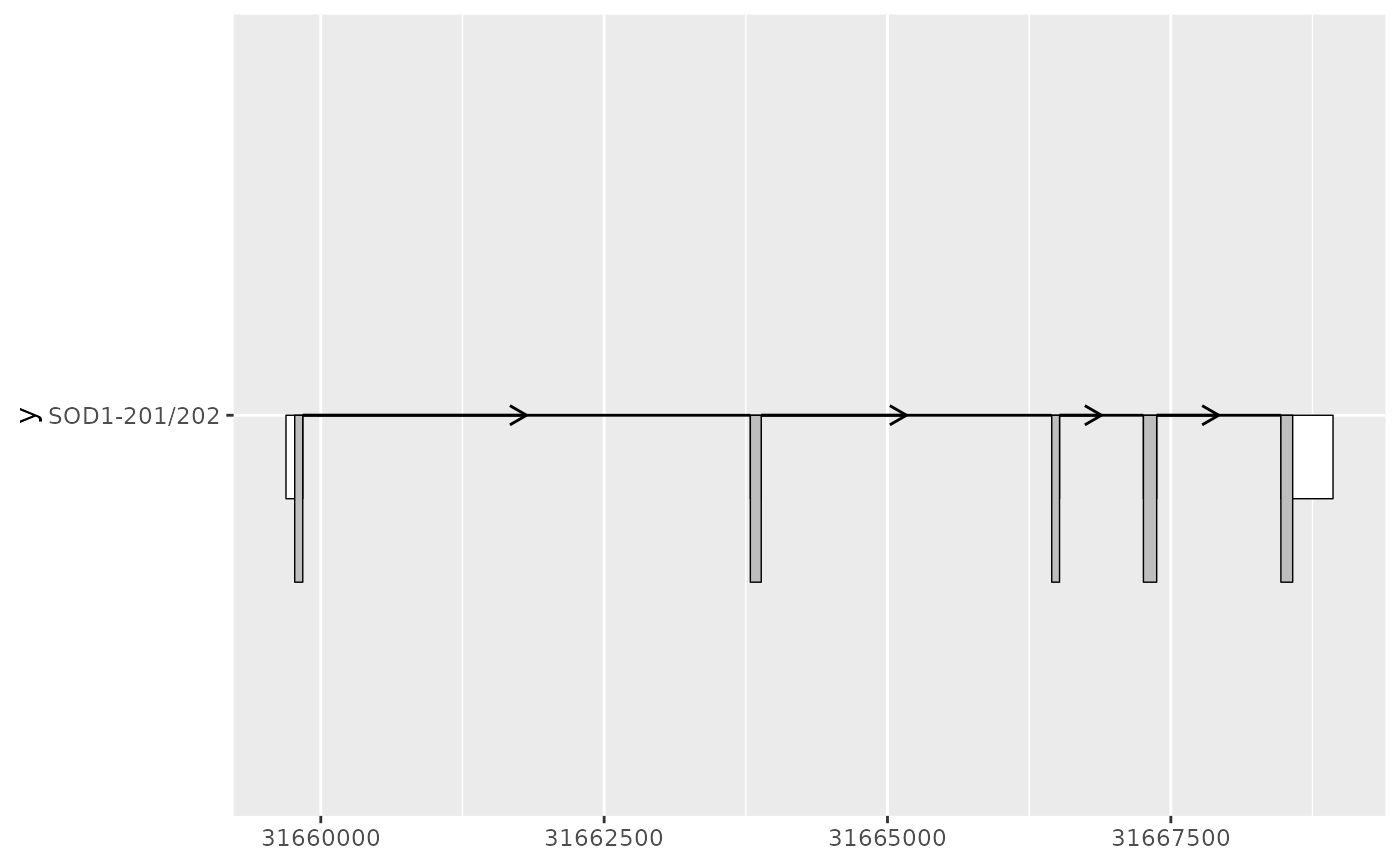
 # geom_half_range() can be useful for comparing between two transcripts
# enabling visualization of one transcript on the top, other on the bottom
sod1_201_exons <- sod1_exons %>% dplyr::filter(transcript_name == "SOD1-201")
sod1_201_cds <- sod1_cds %>% dplyr::filter(transcript_name == "SOD1-201")
sod1_202_exons <- sod1_exons %>% dplyr::filter(transcript_name == "SOD1-202")
sod1_202_cds <- sod1_cds %>% dplyr::filter(transcript_name == "SOD1-202")
sod1_201_plot <- sod1_201_exons %>%
ggplot(aes(
xstart = start,
xend = end,
y = "SOD1-201/202"
)) +
geom_half_range(
fill = "white",
height = 0.125
) +
geom_half_range(
data = sod1_201_cds
) +
geom_intron(
data = to_intron(sod1_201_exons, "transcript_name")
)
sod1_201_plot
# geom_half_range() can be useful for comparing between two transcripts
# enabling visualization of one transcript on the top, other on the bottom
sod1_201_exons <- sod1_exons %>% dplyr::filter(transcript_name == "SOD1-201")
sod1_201_cds <- sod1_cds %>% dplyr::filter(transcript_name == "SOD1-201")
sod1_202_exons <- sod1_exons %>% dplyr::filter(transcript_name == "SOD1-202")
sod1_202_cds <- sod1_cds %>% dplyr::filter(transcript_name == "SOD1-202")
sod1_201_plot <- sod1_201_exons %>%
ggplot(aes(
xstart = start,
xend = end,
y = "SOD1-201/202"
)) +
geom_half_range(
fill = "white",
height = 0.125
) +
geom_half_range(
data = sod1_201_cds
) +
geom_intron(
data = to_intron(sod1_201_exons, "transcript_name")
)
sod1_201_plot
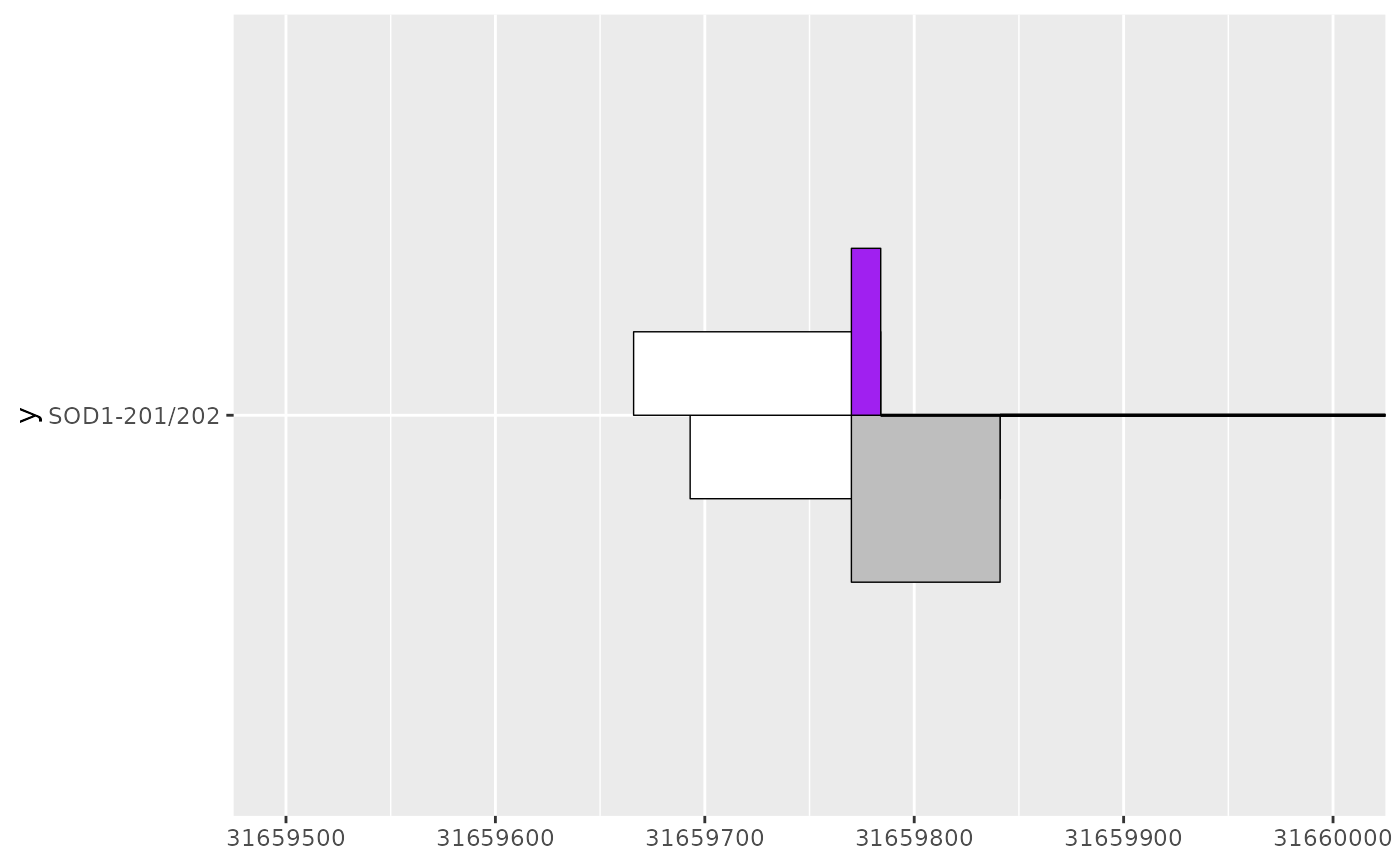
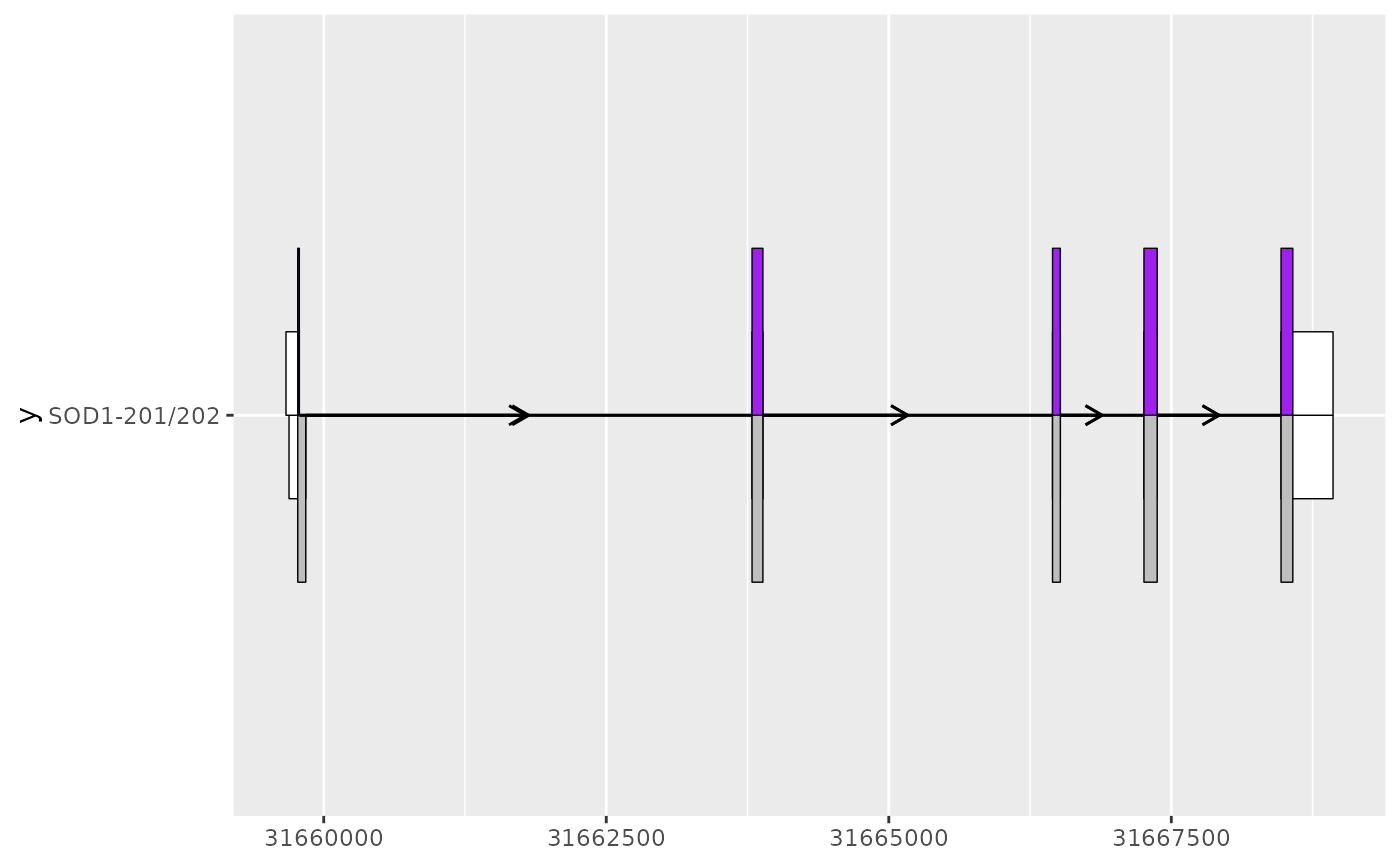
 sod1_201_202_plot <- sod1_201_plot +
geom_half_range(
data = sod1_202_exons,
range.orientation = "top",
fill = "white",
height = 0.125
) +
geom_half_range(
data = sod1_202_cds,
range.orientation = "top",
fill = "purple"
) +
geom_intron(
data = to_intron(sod1_202_exons, "transcript_name")
)
sod1_201_202_plot
sod1_201_202_plot <- sod1_201_plot +
geom_half_range(
data = sod1_202_exons,
range.orientation = "top",
fill = "white",
height = 0.125
) +
geom_half_range(
data = sod1_202_cds,
range.orientation = "top",
fill = "purple"
) +
geom_intron(
data = to_intron(sod1_202_exons, "transcript_name")
)
sod1_201_202_plot
 # leveraging existing ggplot2 functionality via e.g. coord_cartesian()
# can be useful to zoom in on areas of interest
sod1_201_202_plot + coord_cartesian(xlim = c(31659500, 31660000))
# leveraging existing ggplot2 functionality via e.g. coord_cartesian()
# can be useful to zoom in on areas of interest
sod1_201_202_plot + coord_cartesian(xlim = c(31659500, 31660000))